
ICH GUIDELINE FOR PHARMACOVIGILANCE
- Organization and objective of ICH
- Expedited reporting
- Individual case safety reports
- Periodic safety update reports
- Post approval expedited reporting
- Pharmacovigilance planning
- Good clinical practice in pharmacovigilance studies
ORGANIZATION AND OBJECTIVE OF ICH
• ICH is the international council/conference on harmonization of technical requirement for registration of pharmaceuticals for Human Use.

• ICH origin in 1990, and gradually evolved to respond increasingly global developments in pharmaceutical sector.

ICH MEMBER
• Representatives from 6 parties that represent the regulatory bodies and research-based industry in EU, Japan and USA.

ICH ORGANIZATION

1. ICH Steering Committee
• Each of the 6 ICH parties has 2 seats on the ICH steering committee.
• The function of this body includes;
a) Governs ICH
b) Determines polices and has seats on ICH steering
c) Select topic for harmonization
d) Monitors progress of harmonization initiatives.
2. ICH Co-Ordinator
• Helps in monitoring & functioning of ICH.
• The coordinators are nominated by each of the 6 parties.
• Acts as the main contact point with ICH secretariat.
3. ICH Secretariat
• The main function includes;
a) Prepare for documentation of meeting of steering committee.
b) Coordinate preparation for working group and discussion group meeting.
4. ICH working groups
• Steering committee will endorse establishment of three working groups;
a) Expert group (EWG)
b) Implementation working group (IWG)
c) Informal working group
5. ICH operation

SOME IMPORTANT TOPIC CODE
E1: The extended population to assess clinical safety for Drug Intended for Long-Term Treatment of Non-Life Condition.
E2A: Clinical Safety Management: Definitions and standards for Expedited Reporting.
E2B (R2): Maintenance of the clinical safety date management including data elements for transmission of individual case safety reports.
E2B (R3): Clinical safety data management: Data element for transmission of individual case safety reports.
E2C (R1): Clinical safety data management: periodic safety update reports for marketed drugs.
E2C (R2): Periodic benefit-risk evaluation report.
E2D: Post-Approval Safety Data Management: Definitions and standard for expedited reporting.
E2E: Pharmacovigilance Planning
E2F: Developmental safety update report.
EXPEDITED REPORTING

• All adverse drug reaction (ADRs) that are both serious and unexpected are subjected to expedited reporting.
• Any individual case found during clinical trials or individual case safety reports they must be submitted the report to the regulatory authorities within 15-90 days.
• The observer filled a form for this process is known as SUSAR (Suspected Unexpected Serious Adverse Reaction) in the clinical trials
Serious ADR Expedited Reporting
• To report form spontaneous sources and from any type of clinical and epidemiological investigation, independent of design purpose.
• The sponsor or manufacturers report the appropriate data (Serious, severe and causality) to appropriate authority.
Unexpected Adverse Drug Reaction
• The nature or severity of which is not consistent with the applicable product information.
• Some reporting protocols are;
• A company’s investigator Brochure of the medicinal product which is not approved for marketing in the country.
• Investigate the section III F and ICH guidelines in Investigator Brochure of the company.
• To study the report carefully which add the significant information on specific or an expected or unexpected event.
INDIVIDUAL CASE STUDY REPORTS (ICSR)
• The ICSR is defined as an adverse event report for an individual patient and it is also a source of data in the pharmacovigilance.
• It contains ADR report of the individual volunteer in the clinical trials.
• Maintain individual level of report in the WHO database.
COMPONENT OF ICSR
- An identifiable patient
- An identifiable report
- Susceptible drug
- An adverse event
SOURCES OF INDIVIDUAL CASE SAFETY REPORTS
1. UNSOLICITED SOURCE
a) Spontaneous reports
b) Literature
c) Internet
d) Other source
Spontaneous reports
• A spontaneous report is an unsolicited communication by a healthcare professional or consumer to a company, regulatory authority or other organization (e.g., WHO, Regional Center, Poison Control Center) that describes one or more adverse drug reactions in a patient who was given one or more medicinal products and that does not derive from a study or any organized data collection scheme.
Literature
• This is a specific data about the reports which contains systematic literature reviews and reference database for a particular disease or drug in worldwide population.
• The information shared by different local or international journals.
Internet
• It is the biggest platform to gather information regarding adverse drug reactions.
• There are various official websites which shows the correct data about the ADRs.
Other source of data
• Non-medical source like NEWS, Media etc.
• This type of report has some criteria for applying this report.
2. SOLICITED SOURCE
• These types of data are derived from trusted collection system, which are organized by various clinical trials for welfare of people and post approval programs.
3. CONTRACTUAL AGREEMENT FOR DATA
• In this type of data, a contact is necessary between two of the companies which may market the same product in same countries or different regions.
• The main target of these types of arrangements is to avoid duplicate reporting and provide actual report to the regulatory authorities.
4. REGULATORY AUTHORITY SOURCES
• All the serious or unexpected adverse drug reactions and reports are submitted by the marketing authorization holders (MAH) to the regulatory authorities directly.
• The regulatory authorities play an important role for the selection of the reports provided by the MAH.
IMPORTANT INFORMATION FOR MAKING A VALID ICSRS
- Administrative Information
a) Country
b) Type of report
c) Qualification of the reporter
d) Literature reference
- Patient information
a) Patient age
b) Sex
c) Medical history of the patient
d) Seriousness of event
e) Death information
f) Parent child information
3. Reaction information
a) Originality of event or reaction
b) Date of unset of reaction
c) Seriousness or outcomes of reaction
d) Laboratory tests or results
4. Drug information or history (This step includes full description about the drug used by patient with their characterization, trade name, date of stating usage and stop usage of drugs)
a) Drug information with administrative data
b) Drug onset time
c) Dose indication
d) Challenges and Re-Challenges of drug
SUBMISSION OF ICSR’S
• After collaboration with various national and pharmacovigilance centers it is submit via electronic media from any part of the world.
• For submission ICSR’s report the criterial is given in the official site of FDA www.fda.gov.
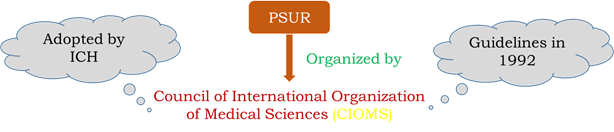
PERIODIC SAFETY UPDATE REPORTS (PSUR)
• The PV documents which are generated for providing an evaluation of risk-benefit (comparison between the risks of a situation and its benefits) balance of a medical product for submission by the marketing authorization holder at define time points during the post authorization phase.
• A PUSR can also contain emerging information of new signals and potential risk of a medicine.
OBJECTIVE
• Summarizes relevant new safety information that may impact the benefit risk profile.
• Examine whether new information is in accord with previous knowledge of the benefit risk profile.
• Summarizes the efficacy and effectiveness.
GENERAL PRINCIPLE
• One report for product containing one active substance authorized to one marketing authorization holder (MAH).

PSUR Content
TITLE
• PSURs contain proprietary information, so the title page should contain a statement on the confidentiality of the data and conclusions included in the report.
EXECUTIVE SUMMARY
• The executive summary should consist of a brief overview providing the reader with a description of the most important information.
INTRODUCTION
• The introduction puts the report in context, describing those products/formulations that are included and excluded, outlining the pharmacology of the product, its indications (both marketed and in clinical trials)
WORLDWIDE MARKETING AUTHORISATION STATUS
• The PSUR should include a short summary of the worldwide marketing authorization status
PSUR Content UPDATE ON REGULATORY AUTHORITY OR MAH ACTIONS TAKEN FOR SAFETY REASONS
• The update on regulatory authority or MAH actions taken for safety reasons refers to marketing authorization, withdrawal or suspension; failure to obtain a marketing authorization renewal; restrictions on distribution; clinical trial suspension; dosage modification/formulation changes and changes in target population or indications.
CHANGES IN REFERENCE SAFETY INFORMATION
• The changes in reference safety information section refers to changes in the CCSI (company core safety information). The CCDS (company core data sheet), which incorporates the CCSI, should be included as an appendix.
PSUR Content PATIENT EXPOSURE
• Patient exposure refers to both market exposure and clinical trials (if relevant). Estimates of patient exposure for marketed drugs often rely on gross approximations of in-house or purchased sales data or volume.
PRESENTATION OF INDIVIDUAL CASE HISTORIES
• There is no specific guidance in E2C (describe the format, content, and timing of a periodic Benefit-Risk Evaluation Report) on the presentation of individual case histories, but because it is impractical to present all case reports for the reporting period, a brief description of the criteria used to select cases for presentation should be given.
• This section of the PSUR should contain a description and analysis of selected cases, including fatalities, presenting new and relevant safety information and grouped by medically relevant headings
PSUR Content STUDIES
• Studies refer to only those company-sponsored studies and published safety studies, including epidemiology studies, that produce findings with potential impact on product safety information.
OTHER INFORMATION
• Other information may include risk management programmes the MAH has put in place and/or a benefit–risk analysis report.
• If such an analysis has been conducted separately, a summary of the analysis should be included in this section.
• This section can also include important information received after the DLP (data lock point).
PSUR Content OVERALL SAFETY EVALUATION
• The overall safety evaluation should highlight new information on serious and non-serious unlisted ADRs. If there are no new safety issues, this should be stated with a note that the information is in keeping with the established safety profile. This section should also review reports of
• drug interactions
• overdose: deliberate or accidental and treatment
• abuse or misuse
• pregnancy or lactation: positive and negative experiences
• special patient groups (e.g., children, elderly, organ impaired)
• effects of long-term treatment
CONCLUSION
• The conclusion should indicate safety data which are not in accordance with previous experience and/or with the CCSI and specify and justify any action recommended or initiated.
POST APPROVAL EXPEDITED REPORTING
• According to good pharmacovigilance practice guidelines all the serious and non-serious ADRs are subjects of expedited reporting.
• Sometimes many ADRs came in post phase of approvals so it’s the responsibility of the MAH that they have to provide knowledge about these reaction to regulatory authorities.
STANDARD OF EXPEDITED REPORTING
• It is easy to choose the right reporting and it’s also easy to differentiate the unexpected reporting and expedited reporting’s.
- Serious ADRs
• The claimed serious reporting is judge by the health care professional and MAH which decides the case is serious or not. If real then it said to be ADRs.
- Observation related to product
• As per GVP guidelines the safety or efficacy study of the medicines produce different statistical date during clinical studies which are helpful in their risk benefit evaluations.
• The drugs which are used in the clinical trials found any life threatening during the studies can be report to the regulatory authorities immediately, so as to save the life of the people.
- Lack of efficacy
• A drug which is not able to produce its effect efficiently.
• If the drugs or product detected as lack of efficacy, then it should be discussed in the periodic safety reports briefly.
4. Overdosing
• Over dosing produce serious disease so it is included in the list of expedited report.
• The regulatory authorities watch the possible outcomes of a medicinal product.
CRITERIA FOR REPORTING OF EXPEDITED REPORTS
• There are some steps which are necessary for the selection of an expedited report.
1. An identifiable reporter
2. An identifiable patient
3. An ADRs
4. A suspect product
HOW TO REPORT
• The forms are available in the official site of CIOMS (Council for international organization of medical sciences).
• The medicinal dictionary for regulatory activity (MedDRA)is used for the coding of medical information.
• Nowadays the e-submission is available for submit the reports.

Hi…!! This is Smrutiranjan Dash, Assistant Professor of Pharmacology from Odisha, India. With a passion for teaching and a dedication to advancing the field of pharmacology, I am committed to sharing knowledge, fostering innovation, and inspiring future healthcare professionals.
Im very pleased to find this site. I need to to thank you for ones time for this particularly fantastic read!! I definitely really liked every part of it and I have you bookmarked to see new information on your site.
Hello my family member! I wіsh to ѕay that this post is
amazing, nice written and include almost all significant infos.
I would like to look extra posts like this .