
Pharmacodynamics
· Pharmacodynamics is derived from the Greek word i.e. “Pharmacon” means Drugs and “Dynamics” means Action.
· It is the study of biochemical and physiological effects of drug and their mechanism of action at organ level and cellular level.
· “What the drugs does to the body when they administered”.
Site of drugs action
· Extracellular
· Cellular
· Intracellular
Principle/types of drugs action
1. Stimulation
· The enhancement of the activity of specialized cells.
· Example: Adrenaline stimulates heart, pilocarpine stimulates salivary glands.
· Excessive stimulation is often followed by depression of that function.
· Example: high dose of picrotoxin (CNS stimulant) produces convulsions followed by coma and respiratory depression.
2. Depression
· The specialized cells selectively diminished.
· Barbiturates depress CNS, quinidine depresses heart, and omeprazole etc. depresses gastric acid secretion.
3. Replacement
· The use of natural metabolites, hormones or their congeners in deficiency states.
· Example: Levodopa in Parkinsonism, insulin in diabetes mellitus, iron in anaemia.
4. Irritation
· Produce noxious effect particularly less specialized cells (epithelium, connective tissue).
· Strong irritation results in inflammation, corrosion, necrosis and morphological damage.
5. Cytotoxic
· Produce Selective cytotoxic action on invading the pathogens or cancerous cells.
· Example: penicillin, chloroquine, zidovudine, cyclophosphamide, etc.
Mechanism action of drugs
· Only few drugs work by their simple physical or chemical properties; examples are.
o Bulk laxatives (ispaghula)—physical mass
o Para amino benzoic acid—absorption of UV rays
o Activated charcoal—adsorptive property
o Mannitol, mag. sulfate—osmotic activity
o 131 I and other radioisotopes—radioactivity
o Antacids—neutralization of gastric HCl
o Pot. permanganate—oxidizing property
o Chelating agents (EDTA, dimercaprol)—chelation of heavy metals.
· Most drugs produce effects by interacting with a different target biomolecule, which usually is a protein.
· Functional proteins that are targets of drug action can be grouped into four major categories i.e.
1. Enzymes
2. Ion channels
3. Transporters
4. Receptors.
1. Enzyme
· Almost all biological reactions are carried out under catalytic influence of enzymes;
· Drugs can either increase or decrease the rate of enzymatically mediated reactions.
Enzyme inhibition
· Some chemicals (heavy metal salts, strong acids and alkalies, formaldehyde, phenol, etc.) denature proteins and inhibit all enzymes non-selectively.
· The selective inhibition of a particular enzyme is a common mode of drug action.
· Such inhibition is either competitive or noncompetitive.
Competitive
Equilibrium type
· The drug is structurally similar, competing with the normal substrate for the enzyme catalytic binding site, which does not create a product or create a non-functional product.
Non-equilibrium type
· The drugs bind with same catalytic site of the enzyme.
· Either formation of covalent bond or have such high affinity for the enzyme that the normal substrate is not able to displace the inhibitor
· Example: Organophosphates react covalently with the esteretic site of the enzyme cholinesterase
Enzyme | Endogenous subtract | Competitive inhibitor |
Cholinesterase | Acetylcholine | Physostigmine, Neostigmine |
Monoamine-oxidase A (MAO-A) | Catecholamines | Moclobemide |
Dopa decarboxylase | Levodopa | Carbidopa, Benserazide |
Xanthine oxidase | Hypoxanthine | Allopurinol |
Angiotensin converting enzyme (ACE) | Angiotensin-1 | Captopril |
5α-Reductase | Testosterone | Finasteride |
Aromatase | Testosterone, Androstenedione | Letrozole, Anastrozole |
Bacterial folate synthase | Para-amino benzoic acid (PABA) | Sulfadiazine |
Non-competitive
· The inhibitor bind to the enzyme adjacent from the active site and loses the catalytic properties.
Non-competitive inhibitor | Enzyme |
Acetazolamide | Carbonic anhydrase |
Aspirin, Indomethacin | Cyclooxygenase |
Disulfiram | Aldehyde |
Omeprazole | H+ K+ ATPase |
Digoxin | Na+ K+ ATPase |
Theophylline | Phosphodiesterase |
Propylthiouracil | Peroxidase in thyroid |
Lovastatin | HMG-CoA reductase |
Sildenafil | Phosphodiesterase-5 |
2. Ion channels
· Ion channels are integral membrane proteins that form a pore to allow the passage of specific ions by passive diffusion.
· Some channels are operated by signal molecules either directly or through G-Protein.
· Directly: Ligand gated channels (e.g. nicotinic receptor)
· G-protein: G-protein regulated channels (e.g. cardiac β1 adrenergic receptor activated Ca2+ channel).
· Drugs can also act on voltage operated and stretch sensitive channels by directly binding to the channel and affecting ion movement through it, e.g. local anaesthetics which obstruct voltage sensitive Na+ channels.
· Certain drugs modulate opening and closing of the channels, e.g.:
o Nifedipine blocks L-type of voltage sensitive Ca2+ channel.
o Ethosuximide inhibits T-type of Ca2+ channels in thalamic neurons.
3. Transporter
· Several subtracts are transported across the membrane by carrier, which facilitate diffusion in the direction of the concentration gradient or pump the metabolite/ion against the concentration gradient using metabolic energy.
· Many drugs are interacting with solute carrier class (SLC) of transporter protein and exert their action by directly.
· Example:
o Norepinephrine transporter (NET): blocked by Desipramine and Cocaine
o Serotonin reuptake transporter (SERT): blocked by Fluoxetine
o dopamine transporter (DAT): blocked by Amphetamines
o vesicular mono-amine transporter (VMAT-2): blocked by Reserpine
o Choline uptake into cholinergic neurons: Hemicholinium.
4. Receptor
· Receptors are defined as cellular macromolecule or binding site located on the surface or inside the effector cell that serves to recognized the signal molecules or drugs.
· Drugs + Receptor – drug receptor complex – Response
Drugs receptor interaction
Selectivity
· Degree of complimentary co relation between drug and receptor
· Example: Adrenaline Selectivity for α, ß Receptor.
Affinity
· Ability of drug to get bound to the receptor.
Intrinsic activity (IA) or Efficacy
· Ability of drug to produce a pharmacological response after making the drug receptor complex.
Agonist
· An agent which activates a receptor to produce an effect similar to that of the physiological signal molecule.
Partial agonist
· An agent which activates a receptor to produce submaximal intrinsic activity (IA between 0 and 1).
· Example: Dichloroisoproterenol (on β adrenergic receptor), pentazocine (on μ opioid receptor).
Inverse agonist
· An agent which activates a receptor to produce an effect in the opposite direction to that of the agonist.
Antagonist
· An agent which prevents the action of an agonist on a receptor or the subsequent response, but does not have any effect of its own.
Competitive antagonist
· The agonist and antagonist binds to the same site of the receptor, they are said to be “Competitive”.
· The efficacy and repulsion depends on the concentration of the agonist and antagonist.
· Drug response curve shifted rightward.
· Example: Acetylcholine = Atropine, Morphine = Naloxone.
Non-competitive antagonist
· It binds to another side of the receptor.
· The response depends on the concentration of the antagonist.
· Drug response curve is flattening.
· Example: Diazepam = Bicuculline
Ligand
· Any molecule which attaches selectively to particular receptors or sites.
Transducer mechanism
1. G – Protein couple receptor (GPCRs)
· The GPCR consist of 7 α – helical membrane spanning hydrophobic amino acid segments which run into 3 extracellular and 3 intracellular loops.
· The GPCRs composed of α, β, and γ subunits.
· The G – protein and their domains laying in the cytosol.
· In the inactive state GDP is bound to the sub-unit at the exposed domain.
· After activation state GTP is split to GDP.
· The activated α subunit carrying GTP dissociate form the other subunits and shows either activation and inhibition effects.
· The β and γ subunits are also shows the operation of K+ channels and inhibit voltage gated Ca2+ channels.
· Action of receptor
o Gs : Adenylyl cyclase activation, Ca2+ channel opening.
o Gi : Adenylyl cyclase inhibition, K+ channel opening.
o Go : Ca2+ channel inhibition.
o Gq : Phospholipase C activation.
· There are three major pathways of GPCRs.
1. Adenylyl cyclase (cAMP pathway)
2. Phospholipase C (IP3 DAG pathway)
3. Channel regulation
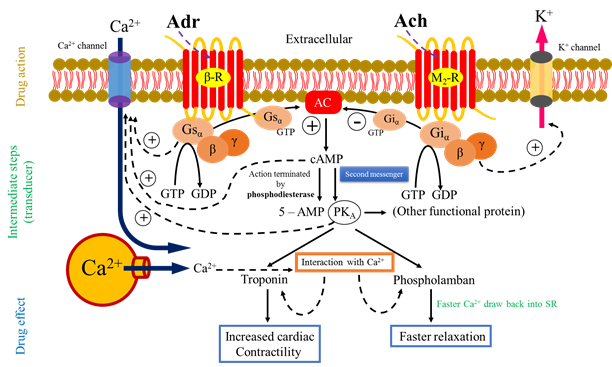
1. Adenylyl cyclase (cAMP pathway)
· After activation of receptor (Adr – binds to β adrenergic receptor) α subunit with GTP activate AC.
· Activation of AC leads to intracellular accumulation of second messenger cAMP that acts primarily through cAMP – dependent protein kinase (PKA).
· The PKA phosphorylates many functional protein including troponin and phospholamban, and alter the function of several enzyme, ion channels, transporter, transcription factor and structural protein to produce;
o Increased contractility (impulse generation – cardiac)
o Glycogenolysis
o Lipolysis
o Hormone synthesis
· Phospholamban and troponin interact with Ca2+, respectively resulting faster relaxation and increased force of contraction.
· The cAMP direct open Ca2+ channel (cyclic nucleotide gated channel – CNG) in heart, brain, and kidney.
· Ach binds to M2 receptor and produce adenylyl cyclase inhibition through inhibitory Gi – protein.
· The action of cAMP is terminated by the enzyme phosphodiesterase (PDEs) which hydrolyzed it to 5 – AMP.
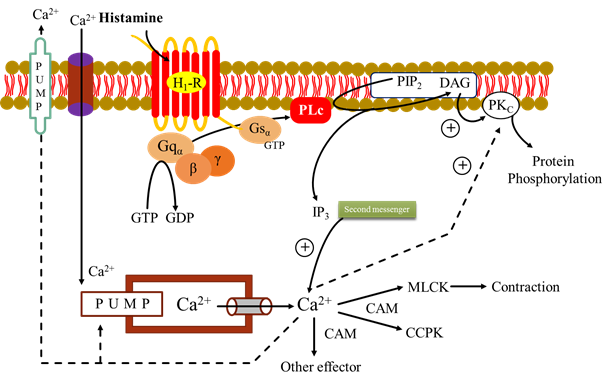
2. Phospholipase C (IP3 – DAG)
· The agonist e.g. histamine binds to its H1 receptor (H1R) and activated α subunit that binds GTP in place of GDP.
· The subunits β and γ are dissociates from activated α subunit.
· Α subunit with GTP activate membrane bound PLc that hydrolyses phophatidyl inositol 4, 5 – biphosphate (PIP2). It generate the second messenger inositol 1, 4, 5 – triphosphate (IP3) and diacylglycerol (DAG).
· The IP3 (water soluble) facilitate Ca2+ mobilization from ER.
· DAG (lipid soluble) conjunction with Ca2+ activates protein kinase C (PKc) which phosphorylates and alter the activity of a number functional and structural protein.
· The cytosolic Ca2+ combines with CAM (calmudulin) to activate myosin light chain kinase (MLCK) inducing contraction. Another regulator calcium calmudulin protein kinase (CCPK).
· Cytosolic Ca2+ is recycled by uptake into the ER as well as effluxes by membrane Ca2+ pump.
3. Channel regulation
· The activated G – proteins (Gs, Gi, and Go) can also open or inhibit ionic channels specific for Ca2+ and K+ without the intervention of any second messenger like CAMP or IP3 and bring about hyperpolarization/depolarization/changes in intracellular Ca2+.
· Gs – Open Ca2+ – myocardium and skeletal muscle.
· Gi, Go – Open K+ – heart and smooth muscle
· Dissociated G protein – (β, γ diamer) – physiological response like Ionotropic, chronotropic, neuronal activity, transmitter release, and smooth muscle relaxant etc.
2. Ion channel receptor
· These cell surface receptors, also called ligand gated ion channels (Na+, K+, Ca2+ or Cl¯).
· Agonist binding opens the channel and causes depolarization/hyperpolarization/changes in cytosolic ionic composition, depending on the ion that flows through.
· Example:
o nicotinic
o cholinergic,
o GABAA,
o glycine (inhibitory AA),
o Excitatory AA-glutamate [kainate, NMDA (N-methyl-D-aspartate receptor) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)] and 5HT3.
3. Transmembrane enzyme linked receptor.
· The third major family of receptor consist of a protein that spans the membrane once and may form dimer or multi sub-unit complexes.
· When these receptor activated, undergoes conformation changes resulting in increased cytosolic enzyme activity.
· This response lasts on the order of minutes to hours.
· The most common enzyme linked receptors
o Epidermal growth factor
o Platelet-derived growth factor
o Atrial natriuretic peptide
o Insulin, and others are those have a Tyrosine kinase activity.
· Binding of the ligand to receptor subunit, undergoes conformational changes to convert inactive to active kinase form.
· After that phosphorylation of tyrosine residue on the specific protein.
Example:
· Tyrosine residues of the β – subunit are auto-phosphorylated.
· Receptor tyrosine kinase phosphorylates other protein e.g. Insulin receptor substrates (IRS).
· Activation of multiple signaling pathways, such as Inositol triphosphate and the Mitogen activated protein kinase (MAP) system.
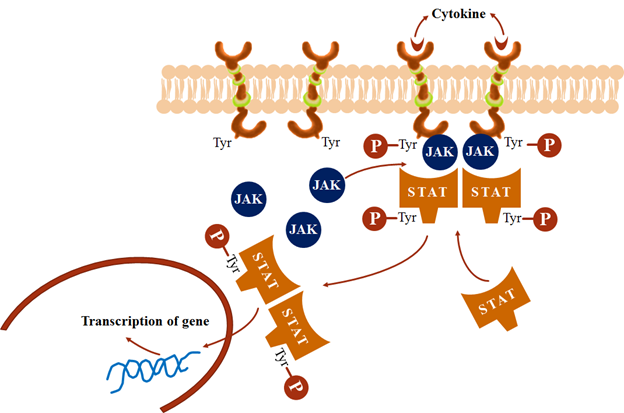
4. Transmembrane JAK-STAT binding receptor
· It has no any intrinsic catalytic domain.
· Cytokines/hormones binding to the extracellular domain induce receptor dimerization which activates the intracellular domain to bind free moving JAK (Janus Kinase) molecules.
· The activated JAK phosphorylate tyrosine residues on the receptor which then binds another protein STAT (signal transducer and activator of transcription).
· Tyrosine residues of STAT also get phosphorylated by JAK.
· The phosphorylated STAT dimerize, dissociate from the receptor and move to the nucleus to regulate transcription of target genes.
· Many cytokines, growth hormone, prolactin, interferons, etc. act through this type of receptor.
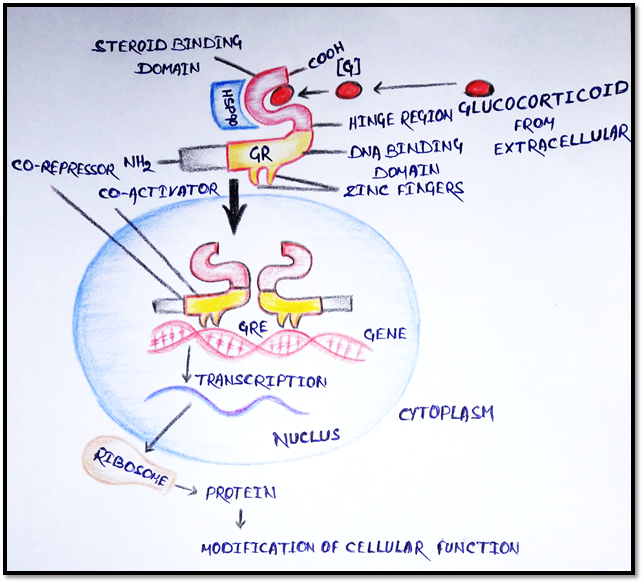
5. Receptor regulating gene expression
· These are intracellular (cytoplasmic or nucleus) soluble proteins which respond to lipid soluble chemical messenger that penetrate the cell.
· Example: Glucocorticoid penetrates the cell membrane and binds to GR protein.
· The GR protein present in cytoplasm with heat shock protein 90 (HSP90), and other protein.
· The GR has a steroid binding domain near the carboxy terminus and mid region DNA binding domain joined by “hinge region”.
· There are two “Zinc fingers” each made up of a loop of amino acid with chelated zinc ion at DNA binding domain.
· Binding of the steroid to GR complex proteins (HSP90) removing their inhibitory action on it.
· Dimerization occurs and steroid bound receptor translocate to the nucleus, binds coactivator/corepressor proteins and interact with specific DNA sequence called “Glucocorticoid responsive elements” (GREs).
· Alteration of gene results promotion (or suppression) of their transcription.
· The specific mRNA translocate the message into a specific protein synthesis, which in turn modifies cell function.
Dose – Response Relationship
· The biological effect of a drug depends on its concentration at the site of action, which in turn is determined by the dose of drug administered, such a relationship is called dose-response relationship.
· When the drug is administered systemically the dose – response relationship has two components
1. Dose-plasma concentration relationship
2. Plasma concentration-response relationship.
A) Graded dose – response relations
· The pharmacological effect increase by increase in the concentration of drug.
· The graded dose response means that the response is continuous.
· The magnitude of the response against increase dose of a drug produce the graded dose-response curve, can be described as a rectangular hyperbola.
1) Potency
· The concentration of drug producing an effect that is 50% of maximum is used to determine potency and is denoted as EC50.
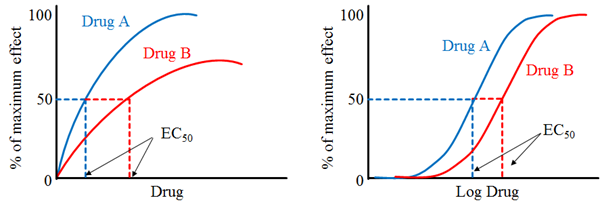
· Drug “A” is more potent than drug “B” because lesser amount of drug “A” is needed, to obtain 50% effect.
· Example: Candesartan is more potent than Irbesartan. Candesartan dose range is 4-32mg whereas Irbesartan dose range is 75-300mg.
· EC50 produce equal response to 50% of the maximal response.
· Smaller the EC50, the more potent of the drug.
2) Efficacy
· It determine from graded dose-response plots is the efficacy of the drug.
· This is the ability of a drug to elicit a response when interacts with a receptor.
· The efficacy is depends on the number of Drug response complexes.
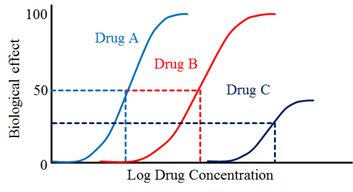
Drug “A” is more potent than drug “B” but both show the same efficacy.
· Drug “C” show lower potency and lower efficacy than drugs “A” and “B”.
B) Effect of drug concentration on receptor binding
· The quantitative relationship between drug concentration and receptor occupancy applied the law of mass action to the kinetic of the binding of drug and receptor molecules.
· Drug + Receptor = Drug – Receptor Complex = Biological effect.
· Mathematically;
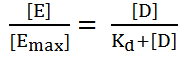
o Where D = the concentration of free drug.
o DR = the concentration of bound drug.
o Rt = the total concentration of receptors and is equal to the sum of the concentration of unbound (free) receptors and bound receptor.
o Kd is used to determine the affinity of drug for its receptor.
o Higher Kd – weaker interaction.
o Lower Kd – higher interaction.
C) Relationship drug binding to pharmacological effect
· Drug concentration and receptor binding can be applied to dose and response.
1. The magnitude of the response is proportional to the amount of receptor bound or occupied.
2. The Emax occur when all receptors are bound.
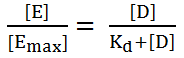
· Where, E = the effect of drug at concentration [D] and Emax = the maximal effect of the drug.

Hi…!! This is Smrutiranjan Dash, Assistant Professor of Pharmacology from Odisha, India. With a passion for teaching and a dedication to advancing the field of pharmacology, I am committed to sharing knowledge, fostering innovation, and inspiring future healthcare professionals.
No matter if some one searches for his necessary thing, so he/she needs to be available that in detail, therefore that thing is maintained
over here.